
Aide
Résultats du docking
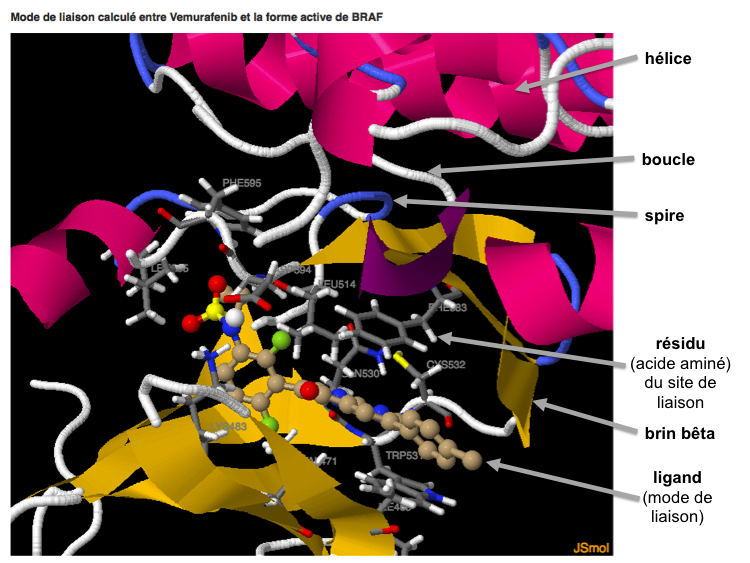
La prédiction du mode de liaison du ligand dans la protéine est donnée sous la forme d'une session 3D dans une nouvelle page (pour les navigateurs testés, dans un nouvel onglet). On peut interagir avec cette session 3D à l'aide d'une souris ou d'un trackpad. Par exemple suivant votre configuration, on peut appliquer des rotations avec le bouton 1 ou faire des zooms avec le bouton 2 (un menu contextuel, peu utile dans notre cas, est obtenu par clic droit).La protéine est représentée en ruban, coloré selon sa structure secondaire: hélices en magenta, feuillets beta en jaune, spires en bleu et boucles en gris. Les résidus de la protéine impliqués dans la liaison avec le ligand sont représentés en bâtonnets, colorés selon la nature des atomes (carbones en gris). Le nom de ces acides aminés est écrit. Le ligand est représenté en boules et bâtonnets, colorés selon la nature des atomes (carbones en bruns-beige).
Plus bas se trouve une représentation graphique de l'échelle des scores, soit la quantification de la liaison du ligand dans la protéine. Plus le score est élevé (vert sur l'échelle), plus la liaison sera forte et donc plus la molécule sera active sur sa cible (thérapeutique ou toxique). Au contraire, une liaison faible, traduisant un mauvais ligand de la protéine, aura un score faible (rouge sur l'échelle). Les scores précalculés pour les molécules existantes sont situés au-dessus de l'échelle, alors que le score calculé provenant du docking d'une nouvelle molécule, est donné numériquement et en dessous de l'échelle.
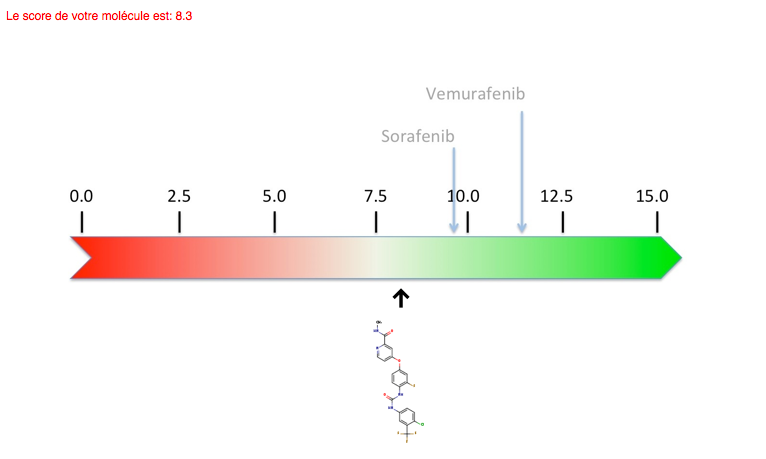
Ceci vous permet de comparer la qualité de la liaison prédite de votre propre molécule "virtuelle" avec des médicaments bien réels.